Обзор новых лекарств 2022 г.
Фарм США. 2022;47(10):24-30.
Новые молекулярные объекты (NME), согласно определению FDA, представляют собой новые лекарственные препараты, содержащие в качестве активного ингредиента химическое вещество, впервые поступающие на рынок в Соединенных Штатах. Следующие описания НМЕ, утвержденные в 2021–2022 гг. ( ТАБЛИЦА 1 ) детализируйте основные клинические и фармакологические профили каждого нового препарата, а также основные меры предосторожности и предупреждения. Также включено краткое изложение отдельных данных по фармакокинетике, побочным реакциям, взаимодействию с лекарствами и дозировке, представленных в FDA в поддержку заявки производителя на новое лекарство. Этот обзор призван быть объективным, а не оценочным по содержанию. Информация для каждого NME была получена в основном из источников, опубликованных до утверждения FDA. Опыт ясно показывает, что многие аспекты терапевтического профиля НМЭ не выявляются в предмаркетинговых клинических исследованиях и проявляются после того, как препарат используется более широкой популяцией. Например, в течение нескольких лет после выхода на рынок некоторых НМЕ становятся очевидными нежелательные реакции, о которых ранее не сообщалось. Некоторые НМЕ могут в конечном итоге получить как минимум одно предупреждение «черного ящика» о серьезных побочных реакциях на лекарства или быть отозваны с рынка по соображениям безопасности, которые не были признаны во время утверждения. Поэтому, хотя этот обзор предлагает введение в некоторые новые препараты, важно, чтобы практикующие врачи знали об изменениях их терапевтических профилей, о которых сообщается в фармацевтической литературе и о которых сообщают пациенты.
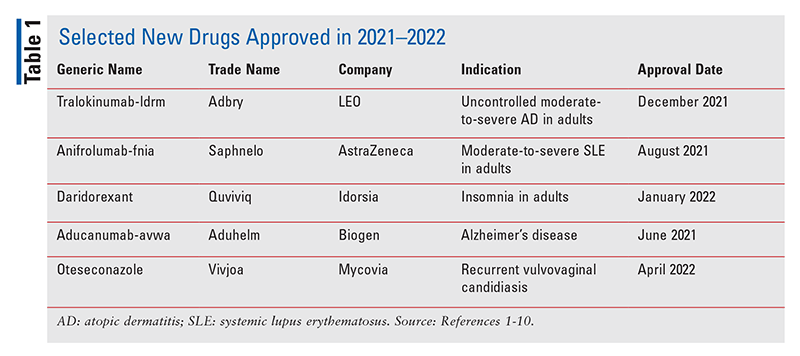
Тралокинумаб-ldrm (Adbry, LEO)
Показания и клинический профиль 1,2 : FDA одобрило тралокинумаб-ldrm для лечения умеренного и тяжелого атопического дерматита (АД) у взрослых, чье заболевание плохо контролируется местными препаратами или когда эти методы лечения не рекомендуются. нашей эры, также известный как атопическая экзема Это хроническое рецидивирующее воспалительное заболевание кожи, характеризующееся сухостью кожи, зудом (особенно ночью) и пятнами от красного до коричневато-серого цвета на различных участках тела. Заболеваемость AD увеличилась почти в три раза в промышленно развитых странах, затрагивая примерно 15-20% детей и 1-3% взрослых во всем мире. Тралокинумаб-ldrm, первый биологический агент, одобренный для лечения АтД, действует путем ингибирования интерлейкина (ИЛ)-13, ключевого медиатора воспаления при этом заболевании. Тралокинумаб-ldrm можно использовать с местными кортикостероидами или без них.
Одобрение этого препарата было основано на результатах трех двойных слепых плацебо-контролируемых многонациональных исследований III фазы (ECZTRA 1, ECZTRA 2 и ECZTRA 3), в которых приняли участие более 1500 взрослых пациентов. Пациенты получали инъекции тралокинумаба-ldrm в дозе 300 мг каждые две недели отдельно или в сочетании с местными кортикостероидами. Первичными конечными точками были общая оценка исследователя 0 или 1 (IGA 0/1) на 16-й неделе и улучшение индекса площади и тяжести экземы на ³75% (EASI 75) на 16-й неделе. Пациенты, достигшие IGA 0/1 и/или EASI 75, получавших тралокинумаб-ldrm на 16-й неделе, были повторно рандомизированы для получения тралокинумаба-ldrm каждые 2 или 4 недели или плацебо на срок до 36 недель. На 16-й неделе больше пациентов, получавших тралокинумаб, чем пациенты, получавшие плацебо, достигли IGA 0/1 (38,9% против 26,2%) и EASI 75 (56,0% против 35,7%). Из респондентов на 16-й неделе 89,6% и 92,5% из тех, кто получал тралокинумаб-ldrm каждые 2 недели, и 77,6% и 90,8% из тех, кто лечился тралокинумаб-ldrm каждые 4 недели, соответственно, сохраняли IGA 0/1 и EASI. 75 на 32-й неделе. Среди пациентов, которые не достигли IGA 0/1 и EASI 75 при применении тралокинумаба-ldrm на 16-й неделе, 30,5% и 55,8% соответственно достигли этих конечных точек на 32-й неделе.
Фармакология и фармакокинетика 1,2 : Тралокинумаб-ldrm представляет собой моноклональное антитело человеческого иммуноглобулина (Ig) G4, которое специфически связывается с человеческим IL-13 и ингибирует его взаимодействие с субъединицами альфа-1 и альфа-2 этого рецептора. IL-13, цитокин, обычно продуцируемый иммунным ответом типа 2, способствует высвобождению провоспалительных цитокинов, хемокинов и IgE, вовлеченных в БА.
Абсолютная биодоступность тралокинумаба-ldrm оценивается в 76% со временем достижения максимальной концентрации в плазме (T Максимум ) от 5 до 8 дней после введения. Объем распределения оценивается в 4,2 л. Тралокинумаб-ldrm, по-видимому, подвергается пептидному метаболизму. Период полувыведения составляет 3 недели, а системный клиренс оценивается в 0,149 л/сут.
Побочные реакции и лекарственные взаимодействия 1,2 : Общие нежелательные явления, о которых сообщалось при применении тралокинумаба-ldrm во время клинических испытаний, включали эозинофилию, воспаление глаз и век, инфекции верхних дыхательных путей и реакции в месте инъекции. Пациентам рекомендуется сообщать лечащему врачу о появлении или ухудшении глазных симптомов, таких как конъюнктивит или кератит. Имели место серьезные реакции гиперчувствительности, включая анафилаксию, и ангионевротический отек, требующие отмены препарата. Tralokinumab-ldrm может снижать эффективность антигельминтной терапии; поэтому его не следует начинать или продолжать у пациентов с ранее существовавшей гельминтозной инфекцией или у тех, кто заразился во время лечения. Во время терапии тралокинумабом следует избегать использования живых вакцин.
Что касается лекарственных взаимодействий, небольшое клиническое исследование по оценке влияния тралокинумаба-ldrm на фармакокинетику мидазолама (субстрат CYP3A4), варфарина (субстрат CYP2C9), омепразола (субстрат CYP2C19), метопролола (субстрат CYP2D6) и кофеина (субстрат CYP1A2) наблюдалось. никаких значительных изменений экспозиции (AUC или C Максимум ) субстратов CYP450 после многократного введения тралокинумаба по сравнению с монотерапией тралокинумабом-ldrm.
Дозировка и администрирование 1,2 : Тралокинумаб-ldrm доступен в предварительно заполненных шприцах на 150 мг/мл для подкожных инъекций. Рекомендуемая доза — начальная доза 600 мг (четыре инъекции по 150 мг), затем 300 мг (две инъекции по 150 мг) каждые две недели. После 16 недель лечения можно рассмотреть дозу 300 мг каждые 4 недели у пациентов с массой тела <100 кг, у которых достигнута чистая или почти чистая кожа. Все соответствующие возрасту прививки, как это рекомендовано действующими руководствами по иммунизации, должны быть выполнены до начала лечения.
Анифролумаб-фния (Сафнело, АстраЗенека)
Показания и клинический профиль 3.4 : Анифролумаб-фния является антагонистом рецепторов интерферона I типа, одобренным для лечения системной красной волчанки (СКВ) средней и тяжелой степени у взрослых, получающих стандартную терапию. Безопасность и эффективность анифролумаба-фнии были установлены в трех многонациональных плацебо-контролируемых клинических исследованиях: MUSE, TULIP-1 и TULIP-2. MUSE было исследованием фазы II, первичные конечные точки которого включали оценку индекса респондеров при СКВ (SRI-4) и устойчивое снижение пероральных кортикостероидов (OCS), измеренное на 24-й неделе. В общей сложности 305 пациентов были рандомизированы 1:1:1 для получения анифролумаба. -фниа 300 мг, анифролумаб-фниа 1000 мг или плацебо. В исследованиях фазы III TULIP-1 и TULIP-2 первичным результатом было улучшение активности заболевания, оцененное через 52 недели, измеренное SRI-4 в TULIP-1 и Композитной оценкой волчанки на основе Британских островов (BICLA) в ТЮЛЬПАН-2. Вторичные результаты TULIP-1 и TULIP-2 включали сохранение снижения OCS, улучшение активности кожной СКВ и частоту обострений.
Показатели ответа BICLA на 52-й неделе были зарегистрированы во всех трех исследованиях, но были первичными конечными точками только в TULIP-2. Разница в частоте ответов составила 28,8% для MUSE, 17% для TULIP-1 и 16,3% для TULIP-2. Частота ответов SRI-4 на 52-й неделе составила 24% для MUSE, 6% для TULIP-1 и 18,2% для TULIP-2. Анифролумаб-фния не продемонстрировал статистически значимой разницы в частоте ответов в TULIP-1.
Фармакология и фармакокинетика 3.4 : Анифролумаб-фния представляет собой моноклональное антитело иммуноглобулина (Ig) G1-каппа, которое связывается с субъединицей 1 рецептора IFN I типа (IFNAR1), ингибируя его активность. Снижение активности ИФН I типа снижает его роль в патогенезе СКВ. Анифролумаб-фния также интернализует IFNAR1, уменьшая количество доступных рецепторов. Эти механизмы действия приводят к уменьшению воспаления и иммунологических процессов, которые способствуют патологии СКВ.
Анифролумаб-фния проявляет нелинейную фармакокинетику в диапазоне доз от 100 мг до 1000 мг каждые 4 недели, что приводит к увеличению воздействия AUC по мере увеличения дозы. Объем распределения у типичного пациента с СКВ (69,1 кг) оценивается в 6,23 л. При стандартной дозировке клиренс анифролумаба-фния 300 мг внутривенно каждые 4 недели оценивается в 0,193 л/сут.
Побочные реакции и лекарственные взаимодействия 3.4 : Наиболее частыми (>10%) побочными реакциями при применении анифролумаба-фния были инфекции (70% пациентов), в том числе грипп, инфекции мочевыводящих путей, туберкулез, бронхит и (34% случаев) инфекции верхних дыхательных путей (например, назофарингит, фарингит). и синусит). Частота серьезных инфекций во время лечения составила 5%. В настоящее время данные об использовании анифролумаба-фнии при беременности человека ограничены и недостаточны для информирования о связанном с препаратом риске серьезных врожденных дефектов, выкидыша или неблагоприятных исходов для матери или плода. Известно, что моноклональные антитела IgG активно транспортируются через плаценту по мере развития беременности; поэтому воздействие анифролумаба-фнии на плод может быть выше в третьем триместре.
Единственным противопоказанием к применению анифролумаба-фнии является тяжелая гиперчувствительность (например, анафилаксия) к анифролумабу или любому компоненту его состава. К клинически заметным побочным реакциям, вызывающим озабоченность, относятся серьезные инфекции, реакции гиперчувствительности, включая анафилаксию, и злокачественные новообразования. На сегодняшний день официальных исследований лекарственного взаимодействия не проводилось.
Дозировка и администрирование 3.4 : Анифролумаб-фния поставляется в виде однодозового флакона, содержащего 300 мг/2 мл (150 мг/мл). Рекомендуемая доза составляет 300 мг в виде внутривенной инфузии в течение 30 минут. Перед введением анифролумаб-фния необходимо развести 0,9% раствором хлорида натрия для инъекций, USP, с использованием асептической техники. 2 мл 0,9% раствора хлорида натрия для инъекций отбирают из инфузионного пакета объемом 100 мл и добавляют 2 мл анифролумаба-фнии при осторожном перемешивании, чтобы общий объем равнялся 100 мл. Раствор для инфузий следует вводить сразу после приготовления в течение 30 минут через инфузионную систему, содержащую стерильный 0,2- или 0,22-микронный встроенный фильтр с низким связыванием белков. После завершения инфузии линию необходимо промыть 25 мл 0,9% раствора хлорида натрия для инъекций, USP.
Даридорексант (Кувивик, Идорсия)
Показания и клинический профиль 5.6 : Даридорексант был одобрен для лечения взрослых с бессонницей, характеризующейся трудностями с засыпанием и/или поддержанием сна. Бессонница, наиболее распространенное нарушение сна (отмечается более чем у 30% взрослых в США), определяется как трудности с получением достаточного количества сна и неудовлетворенность сном в сочетании со значительным негативным влиянием на дневную функцию. Хронической бессонницей считают трудности с началом и/или поддержанием сна не менее 3 ночей в неделю в течение не менее 3 месяцев. Бессонница, которая, по-видимому, возникает в результате гиперактивности областей мозга, связанных с бодрствованием, оказывает значительное влияние на настроение больного, уровень энергии и способность концентрироваться. Кроме того, длительная бессонница связана с серьезными заболеваниями, такими как психические расстройства, сердечно-сосудистые заболевания, диабет 2 типа, злоупотребление психоактивными веществами и деменция.
Эффективность даридорексанта оценивалась в двух рандомизированных многоцентровых плацебо-контролируемых исследованиях: исследование 1 и исследование 2. В общей сложности 1854 субъекта с бессонницей, как определено Диагностическое и Статистическое Руководство по Психическим Расстройствам , 5-е издание, получали даридорексант или плацебо один раз в день вечером в течение 3 месяцев. В исследовании 1 930 субъектов получали даридорексант в дозе 25 мг, даридорексант в дозе 50 мг или плацебо; В исследовании 2 924 субъекта получали даридорексант в дозе 25 мг, даридорексант в дозе 10 мг или плацебо. В конце 3-месячного периода лечения оба исследования включали 7-дневный период прекращения приема плацебо, после которого испытуемые могли участвовать в расширенном исследовании. В общей сложности 600 субъектов лечились в течение не менее 6 месяцев; из них 373 лечились не менее 12 месяцев. Первичными конечными точками эффективности для обоих исследований были изменение от исходного уровня до 1 и 3 месяцев латентного периода к постоянному сну (LPS, время индукции сна) и пробуждения после начала сна (WASO, поддержание сна), объективно измеренные с помощью полисомнографии в лаборатории сна. В исследовании 1 даридорексант в дозе 25 мг и даридорексант в дозе 50 мг продемонстрировали статистически значимое улучшение по сравнению с плацебо уровней LPS и WASO, а также общего времени сна (sTST) по самооценке на 1-м и 3-м месяцах. Доза 50 мг также показала значительное снижение дневного времени сна. сонливость по сравнению с плацебо — по оценке домена сонливости из анкеты 7 симптомов и последствий бессонницы в дневное время — на 1-м и 3-м месяцах, ключевая вторичная конечная точка. В исследовании 2 доза 25 мг показала статистически значимое улучшение по сравнению с плацебо показателей WASO и sTST через 1 и 3 месяца, а доза 10 мг — нет.
Фармакология и фармакокинетика 5.6 : Орексин А и орексин В представляют собой нейропептиды, секретируемые нейронами, преимущественно в латеральном гипоталамусе. Эти нейропептиды проявляют свои эффекты путем связывания и активации двух рецепторов, связанных с G-белком (GPCR): рецептора орексина типа 1 и рецептора орексина типа 2 (OX1R, OX2R). Пути рецепторов орексина играют жизненно важную регулирующую роль во многих физиологических процессах, включая ритм сна и бодрствования. Даридорексант ( ФИГУРА 1 ) представляет собой бензимидазол-пирролидин, предназначенный для функционирования в качестве двойного антагониста OX1R и OX2R, тем самым подавляя стимулирующую бодрствование активность орексина А и орексина В.

Биодоступность даридорексанта при пероральном приеме составляет 62%, а пиковые концентрации в плазме достигаются в течение 1–2 часов (T). Максимум ). Даридорексант имеет объем распределения 31 л и на 99,7% связывается с белками плазмы. Даридорексант подвергается экстенсивному метаболизму, главным образом, CYP3A4 (89%). Основной путь экскреции - через фекалии (57%); меньшая часть выводится с мочой (28%), преимущественно в виде метаболитов. Конечный период полувыведения даридорексанта составляет примерно 8 часов. Быстрое всасывание даридорексанта в начале сна, а также профиль его клиренса, приводящий к 80% выведению после ночного сна, могут минимизировать остаточные эффекты.
Побочные реакции и лекарственные взаимодействия 5.6 : Наиболее частыми побочными реакциями, связанными с даридорексантом в клинических исследованиях (> 5% и выше, чем у плацебо), были головная боль и сонливость или утомляемость. На этикетке продукта также есть предупреждение об усугублении депрессии и суицидальных мыслях, а также о сложном поведении во сне, включая лунатизм, вождение автомобиля во сне и участие в других действиях в состоянии неполного бодрствования (например, приготовление / прием пищи, телефонные звонки, секс). . Пациенты должны немедленно связаться со своим лечащим врачом, если они испытывают какие-либо из этих побочных эффектов. Галлюцинации и сонный паралич (временная неспособность двигаться или говорить) продолжительностью до нескольких минут могут возникать, когда больной засыпает или просыпается. Даридорексант противопоказан пациентам с нарколепсией.
Отсутствуют данные о применении даридорексанта беременными женщинами для определения риска серьезных врожденных дефектов, выкидыша или других неблагоприятных исходов для матери или плода, связанных с приемом препарата. В исследованиях репродукции животных даридорексант не вызывал токсического воздействия на плод в дозах, в 8–10 раз превышающих максимальную рекомендуемую дозу для человека. Нет данных о присутствии даридорексанта в грудном молоке, влиянии на младенцев, находящихся на грудном вскармливании, или влиянии на выработку молока, но препарат и его метаболиты были обнаружены в молоке кормящих грызунов. Таким образом, младенцы, потенциально подвергающиеся воздействию даридорексанта через грудное молоко, должны находиться под наблюдением на предмет чрезмерного седативного эффекта. Даридорексант находится под федеральным контролем, потому что им можно злоупотреблять и это может привести к зависимости.
Дозировка и администрирование 5.6 : Даридорексант выпускается в виде таблеток, покрытых пленочной оболочкой, по 25 и 50 мг для приема внутрь. Рекомендуемая доза составляет от 25 мг до 50 мг один раз в сутки за 30 минут до сна и за 7 часов до запланированного пробуждения. Время засыпания может быть отсрочено, если даридорексант принимается во время или вскоре после еды. Даридорексант не следует принимать с другими депрессантами центральной нервной системы или алкоголем. Пациентам также следует рекомендовать избегать вождения, работы с тяжелыми механизмами или выполнения любой деятельности, требующей ясного мышления, в то время как все еще ощущаются эффекты препарата. У пациентов с нарушением функции печени средней степени тяжести максимальная рекомендуемая доза составляет 25 мг не чаще одного раза за ночь. Даридорексант не рекомендуется применять у пациентов с тяжелой печеночной недостаточностью. Следует избегать одновременного применения даридорексанта с сильными ингибиторами CYP3A4, а дозу следует ограничить до 25 мг у пациентов, одновременно получающих умеренные ингибиторы CYP3A4. Также следует избегать одновременного применения даридорексанта с умеренными или сильными индукторами CYP3A4.
Адуканумаб-авва (Адухельм, Биоген)
Показания и клинический профиль 7,8 : Адуканумаб-авва представляет собой антитело, направленное против бета-амилоида (АВ), показанное для лечения болезни Альцгеймера, и это первый новый препарат для лечения этого заболевания с 2003 года. терапевтическое преимущество перед существующими методами лечения серьезного или опасного для жизни заболевания. Дальнейшее одобрение этого показания зависит от подтверждения клинической пользы в подтверждающих исследованиях. Болезнь Альцгеймера, необратимое, изнурительное и прогрессирующее заболевание головного мозга, поразившее 6,2 миллиона американцев, медленно разрушает память, мышление и, в конечном итоге, способность выполнять даже простые задачи. Хотя то, что именно вызывает ее, до конца не известно, болезнь Альцгеймера характеризуется изменениями в головном мозге, включая амилоидные бляшки и нейрофибриллярные (тау) клубки, которые приводят к потере нейронов в критических областях мозга, регулирующих память и мышление. Доступные в настоящее время методы лечения лечат только симптомы заболевания, но адуканумаб-авва является первым агентом, который воздействует на предполагаемый процесс, лежащий в основе болезни Альцгеймера, и изменяет его.
Ускоренное одобрение адуканумаба-авва было основано на результатах трех двойных слепых плацебо-контролируемых исследований с диапазоном доз, в которых приняли участие в общей сложности 3482 пациента с легкими когнитивными нарушениями или легкой стадией деменции при болезни Альцгеймера. В исследованиях, известных как EMERGE (исследование 1), ENGAGE (исследование 2) и PRIME (исследование 3), использовали позитронно-эмиссионную томографию для количественного определения бляшек AB в областях мозга, вовлеченных в патологию болезни Альцгеймера, по сравнению с областями мозга, не вовлеченными в болезнь. У пациентов, получавших адуканумаб-авва, наблюдалось значительное дозозависимое и зависящее от времени уменьшение бляшек AB (59–71 % в исследованиях), тогда как в контрольной группе уменьшение бляшек AB не наблюдалось. На сегодняшний день нет данных о безопасности или эффективности начала лечения адуканумабом-авва на ранних или поздних стадиях болезни Альцгеймера.
Фармакология и фармакокинетика 7,8 : Адуканумаб-авва представляет собой моноклональное антитело иммуноглобулина-гамма (IgG) 1 человека, которое способно преодолевать гематоэнцефалический барьер для селективного нацеливания и связывания агрегированных растворимых олигомеров и конформаций нерастворимых фибрилл бляшек AB в головном мозге. Биохимические исследования показали, что адуканумаб-авва избирательно связывается с линейным эпитопом, образованным аминокислотами АВ с 3 по 7, тем самым сводя к минимуму взаимодействие с другими белками. На основании слабой одновалентной аффинности, быстрой кинетики связывания и сильной авидности к агрегатам, богатым эпитопами, было показано, что адуканумаб-авва различает мономеры АВ и олигомерные или фибриллярные агрегаты.
Равновесные концентрации адуканумаба-авва достигаются через 16 недель повторного введения дозы каждые 4 недели. Пиковая концентрация (С Максимум ), минимальная концентрация (C мин ), и площадь под кривой зависимости концентрации в плазме от времени в равновесном состоянии, увеличивая дозу пропорционально стандартному диапазону доз (1–10 мг/кг каждые 4 недели). Предполагаемый объем распределения в равновесном состоянии составляет 9,6 л. Предполагается, что подобно эндогенному IgG адуканумаб-авва гидролизуется до небольших пептидов и, в конечном итоге, до аминокислот посредством белкового катаболизма. Конечный период полувыведения адуканумаба-авва составляет около 25 дней. Не сообщалось об исследованиях, оценивающих фармакокинетику у пациентов с почечной или печеночной недостаточностью, но не ожидается, что препарат подвергается выведению почками или метаболизму в печени. Различия в массе тела, возрасте, поле и расе могут привести к изменению воздействия препарата, но не было обнаружено значительного влияния на клинический ответ.
Побочные реакции и лекарственные взаимодействия 7,8 : Наиболее часто сообщаемые нежелательные реакции на адуканумаб-авва включают нарушения визуализации, связанные с амилоидами (АВСА); Головная боль; падает; диарея; спутанность сознания, дезориентация и измененное психическое состояние. На этикетке продукта есть предупреждение об АВСА, которое чаще всего проявляется в виде временного отека частей мозга; этот эффект обычно проходит со временем и, как известно, не вызывает симптомов. Однако некоторые пациенты с АВСА сообщали о головной боли, спутанности сознания, головокружении, изменении зрения или тошноте, связанных с приемом адуканумаба-авва. В редких случаях применение адуканумаба-авва ассоциировалось с реакциями гиперчувствительности, включая ангионевротический отек и крапивницу.
Дозировка и администрирование 7,8 : Адуканумаб-авва поставляется в виде не содержащего консервантов стерильного раствора для внутривенного введения в однодозовых флаконах по 170 мг/1,7 мл (100 мг/мл) и 300 мг/3 мл (100 мг/мл). После первоначального титрования (более 6 недель) рекомендуемая доза составляет 10 мг/кг в виде внутривенной инфузии в течение примерно 1 часа каждые 4 недели с интервалом не менее 21 дня. Первоначальное титрование включает введение доз 1 мг/кг для инфузий 1 и 2; дозы 3 мг/кг для инфузий 3 и 4; и дозы 6 мг/кг для инфузий 5 и 6. Коррекция дозы при почечной или печеночной недостаточности не предполагается.
Отесеконазол (Vivjoa, Mycovia)
Показания и клинический профиль 9.10 : Отесеконазол является первым и единственным препаратом, специально предназначенным для снижения частоты рецидивирующего вульвовагинального кандидоза (РВВК) у женщин с РВВК в анамнезе, не обладающих репродуктивным потенциалом. Его применение противопоказано беременным и кормящим женщинам. РВВК, широко известный как хроническая дрожжевая инфекция, определяется как три или более симптоматических острых эпизода дрожжевой инфекции в течение 12-месячного периода. Симптомы включают вагинальный зуд, жжение, раздражение и воспаление, а у некоторых пациентов наблюдаются аномальные выделения из влагалища и болезненное мочеиспускание или половой акт. Подсчитано, что 75% взрослых женщин хотя бы раз в жизни перенесут дрожжевую инфекцию, и примерно у 50% будут рецидивы; из этих женщин до 90% разовьется РВВК. Управление по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA) присвоило отесеконазолу статус Fast-Track и квалифицированный продукт для лечения инфекционных заболеваний.
Отесеконазол был одобрен на основании данных об эффективности трех клинических испытаний III фазы, включавших 875 пациентов из 232 центров в 11 странах. В двух глобальных исследованиях VIOLET у 93-96% женщин с РВВК, получавших отеконазол, не было рецидивов в течение 48-недельного поддерживающего периода; напротив, от 57% до 61% реципиентов плацебо не имели рецидивов. В проведенном в США исследовании ultraVIOLET у 89,7% женщин с РВВК, получавших отесеконазол, исчезла первичная дрожжевая инфекция и не было рецидивов в течение 50-недельного поддерживающего периода, тогда как у 57,1% женщин, получавших флуконазол и плацебо, рецидивов не было.
Фармакология и фармакокинетика 9.10 : Отесеконазол ( ФИГУРА 2 ) представляет собой новое противогрибковое производное тетразола, которое ингибирует 14-альфа-деметилазу (CYP51), грибковый фермент, который катализирует ранний этап биосинтеза незаменимого грибкового стерола эргостерола. Кроме того, ингибирование 14-альфа-деметилазы приводит к накоплению 14-метилированных стеролов, токсичных для грибов. Из-за включения в свою структуру металлсвязывающей группы тетразола отсеконазол имеет пониженное сродство к ферментам CYP человека и, предположительно, лучшие профили побочных реакций и лекарственного взаимодействия.

Время достижения пиковых концентраций отеконазола в плазме составляет от 5 до 10 часов. Прием пищи с высоким содержанием жиров и калорий увеличивает C Максимум и AUC 0-72ч на 45% и 36% соответственно, но никаких существенных различий не наблюдалось при приеме нежирной и низкокалорийной пищи. Объем распределения отесеконазола составляет приблизительно 423 л, и препарат >99% связывается с белками плазмы. Исследования на животных показывают, что экспозиция отеконазола в тканях влагалища сравнима с экспозицией в плазме. Отесеконазол не подвергается значительному метаболизму. Приблизительно 56% пероральной дозы выводится с калом с желчью, а 26% выводится с мочой. Средний конечный период полувыведения отесеконазола составляет примерно 138 дней.
Побочные реакции и лекарственные взаимодействия 9.10 : Наиболее частыми побочными реакциями были головная боль и тошнота, которые наблюдались у 7,4% и 3,6% участников исследования соответственно. Побочные реакции, о которых сообщалось у <2% пациентов в клинических исследованиях, включали диспепсию, приливы, дизурию, нарушения репродуктивной системы (меноррагию, метроррагию, вульвовагинальное раздражение) и повышение уровня креатинфосфокиназы в крови. В исследованиях на животных было обнаружено, что отесеконазол наносит вред плоду; поэтому он противопоказан женщинам с репродуктивным потенциалом, а также беременным и кормящим женщинам из-за потенциального риска для плода или младенца, находящегося на грудном вскармливании. Отесеконазол также противопоказан пациентам с известной гиперчувствительностью к препарату.
Отесеконазол является ингибитором белка резистентности рака молочной железы (BCRP), и одновременное применение с субстратами BCRP, такими как розувастатин, может увеличить экспозицию субстрата, увеличивая риск побочных реакций, связанных с этими агентами. Таким образом, рекомендуется использовать самые низкие эффективные начальные и поддерживающие дозы субстрата BCRP, если он применяется одновременно с отеконазолом, и следует контролировать состояние пациента на предмет побочных реакций.
Дозировка и администрирование 9.10 : Отесеконазол выпускается в виде твердых желатиновых капсул по 150 мг для приема внутрь. Существует два рекомендуемых режима дозирования: 1) только отесеконазол и 2) отесеконазол плюс флуконазол. На этикетке продукта приведены подробные графики дозирования для этих режимов. Коррекция дозы не рекомендуется пациентам с почечной недостаточностью легкой и средней степени тяжести или легкой печеночной недостаточностью; однако отесеконазол не рекомендуется назначать пациентам с тяжелой почечной недостаточностью или терминальной стадией почечной недостаточности (с диализом или без него), а также пациентам с умеренной или тяжелой печеночной недостаточностью.
ИСПОЛЬЗОВАННАЯ ЛИТЕРАТУРА
1. Вкладыш в упаковку Adbry (тралокинумаб-ldrm). Мэдисон, Нью-Джерси: LEO Pharma Inc; июль 2022.
2. Волленберг А., Хауэлл М.Д., Гуттман-Ясский Э. и соавт. Лечение атопического дерматита тралокинумабом, mAb против IL-13. J Аллергия Клин Иммунол . 2019;143(1):135-141.
3. Листок-вкладыш сафнело (анифролумаб-фниа). Уилмингтон, Делавэр: AstraZeneca Pharmaceuticals LP; июль 2022.
4. Анифролумаб: информация о препарате. Своевременно. Уолтем, Массачусетс: UpToDate; 2022. www.uptodate.com. Accessed June 28, 2022.
5. Вкладыш в упаковку Quviviq (даридорексант). Рэднор, Пенсильвания: Idorsia Pharmaceuticals US Inc; апрель 2022.
6. Маркхэм А. Даридорексант: первое одобрение. Наркотики . 2022;82(5):601-607.
7. Вкладыш в упаковку Адугельма (адуканумаб-авва). Кембридж, Массачусетс: Biogen Inc; июнь 2021.
8. Севиньи Дж., Чиао П., Уильямс Л. и др. Адуканумаб (BIIB037), моноклональное антитело против бета-амилоида, у пациентов с продромальной или легкой формой болезни Альцгеймера: промежуточные результаты рандомизированного двойного слепого плацебо-контролируемого исследования фазы 1b. Альцевая деменция . 2015; 11 (7 доп. 6): P277.
9. Листок-вкладыш Vivjoa (отесеконазол). Дарем, Северная Каролина: Mycovia Pharmaceuticals, Inc; апрель 2022.
10. Sobel JD, Nyirjesy P. Отсеконазол: прогресс в лечении рецидивирующего вульвовагинального кандидоза. Будущая микробиология . 2021;16(18):1453-1461.
Содержание, содержащееся в этой статье, предназначено только для информационных целей. Содержимое не предназначено для замены профессиональной консультации. Вы полагаетесь на любую информацию, представленную в этой статье, исключительно на свой страх и риск.